

Auch kleine Fehler können im Umfeld der Entwicklung, Produktion und Vertrieb von pharmazeutischen Produkten verheerende Wirkung haben. Eine der bekanntesten Katastrophen ist zum Beispiel mit dem Namen Contergan verbunden. Es gibt aber auch andere, weniger bekannte Probleme, die ebenfalls Gesundheit und Leben von Patienten gefährden und die von Fehlern in computergestützten Systemen verursacht werden können.
Der Transport von Impfstoffen wird durch IT-Systeme überwacht. Ein Impfstoff darf zum Beispiel während seines Weges von der Produktion über die Zwischenlager bis zum Endverbraucher nur eine bestimmte Zeitspanne einer Temperatur außerhalb des zugelassenen Temperaturbereichs ausgesetzt werden. Ansonsten führt die Impfung nicht zum gewünschten Impfschutz.
Es gibt kaum Geschäftsprozesse, die ohne Computer auskommen. Computersysteme, die für kritische Aufgaben in der Pharmaindustrie benutzt werden, müssen ganz besonderen Richtlinien genügen, um die Gesundheit und das Leben des Patienten zu schützen.
Im Folgenden werden die Besonderheiten im Lebenszyklus von computergestützten Systemen skizziert und erklärt, worauf man achten muss, damit diese „fit für den Gebrauch“ in pharmazeutischen Geschäftsprozessen sind.
Good Practices – was dahinter steht
Im Pharmaumfeld spricht man oft von GxP-kritischen Geschäftsprozessen oder verkürzt von GxP-Prozessen [ISACA]. Im Akronym GxP steht dabei G für Good und P für Practices. Der Buchstabe „x“ ist ein Platzhalter für Laboratory, Clinical, Manufacturing, Distribution oder Pharmacovigilance. Diese Practices decken das Leben eines pharmazeutischen Produktes von der Entwicklung im Labor bis zur Lieferung an den Patienten ab und sind Richtlinien, die von den zuständigen Behörden festgelegt werden. Sie beschreiben, was man tun muss, um die erforderliche Mindestqualität zu garantieren.
Zuständige Behörden sind zum Beispiel EMA (European Medicines Agency) oder FDA (Food and Drug Administration) für Firmen, die ihre Produkte in den US vermarkten. In Deutschland kommt noch das Bundesinstitut für Arzneimittel und Medizinprodukte „BfarM“ und das Bundesministerium für Gesundheit (BMG) hinzu, sowie für eventuelle weitere Märkte, die beliefert werden, die jeweiligen Ämter (z. B. MHRA für UK). Die GxP enthalten dann ihrerseits wieder mehr oder weniger detaillierte Anweisungen, denen computergestützte Systeme genügen müssen.
Computergestützte Systeme
Ein wichtiges Konzept innerhalb der GxP-Prozesse ist das computergestützte System (siehe Abbildung 1). Solch ein System besteht sowohl aus dem Computer als auch aus Komponenten, die für den Geschäftsprozess von Bedeutung sind.
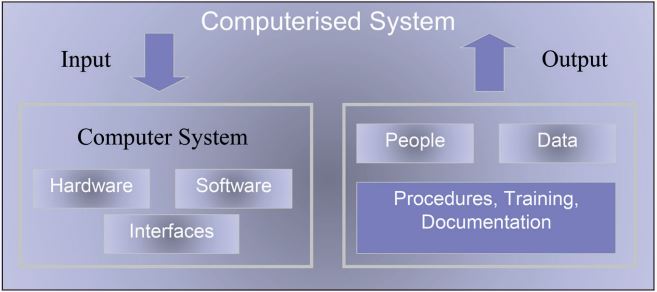
Abb. 1: Computergestütztes System
Also nicht nur Hardware, Software und Daten, die in und aus dem System fließen, sondern auch Benutzer, einschließlich privilegierter Benutzer, wie Systemverwalter. Wichtig ist, dass diese Benutzer in ihrem Profil den Nachweis haben, dass sie durch geeignetes Training für ihre Tätigkeit qualifiziert sind. Das gilt natürlich auch für Supplier von IT-Services.
Genauso wichtig wie das Training der Personen ist auch die Systemdokumentation in Form von SOPs (Standard Operation Procedures), die definieren, wie das computergestützte System für den entsprechenden Geschäftsprozess zu benutzen ist.
Validierung computergestützter Systeme
Regulierungen der Pharmaindustrie und die GxP schreiben vor, dass computergestützte Systeme abhängig von ihrem Risiko für
- die Gesundheit des Patienten,
- die Qualität des Produktes und
- die Integrität der regulierten Daten
validiert werden müssen. Es gibt zahlreiche Definitionen von Validierung, in einem Satz formuliert:
Validierung ist ein kontinuierlicher Prozess, der dokumentierte Beweise erbringt, dass ein computergestütztes System die vorher spezifizierten Anforderungen reproduzierbar im praktischen Einsatz erfüllt.
Nützlich für das Arbeiten im validierten Umfeld ist das Konzept des Lebenszyklus von computergestützten Systemen (siehe Abbildung 2). Dieser Lebenszyklus beschreibt alle Aktionen von der Planung des Systems bis zu dessen „Entsorgung“.
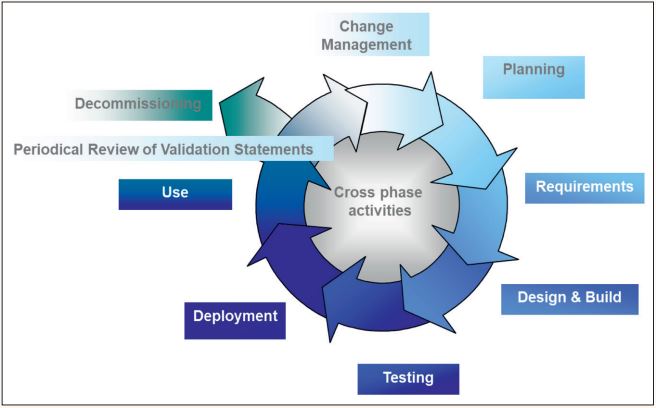
Abb. 2: Lebenszyklus eines computergestützten Systems
Es ist sinnvoll, nicht allen Systemen die gleiche Aufmerksamkeit zu schenken, sondern sich besonders auf diejenigen zu konzentrieren, die ein Risiko darstellen. Dies bedeutet konkret, dass im Prozess der Validierung auch eine Risikobewertung stattfinden muss.
Klassischerweise wird das Risiko durch zwei Faktoren bewertet, die Konsequenz und die Wahrscheinlichkeit, mit der solch ein Problem auftritt (siehe Abbildung 3). Aus diesen beiden Faktoren ergibt sich dann eine Bewertung des Risikos.
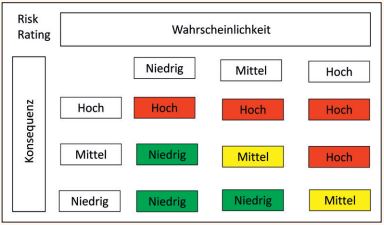
Abb. 3: Risk Matrix – Wahrscheinlichkeit und Konsequenz
Man unterscheidet im Allgemeinen zwei verschiedene Arten von Risiken. Das erste ist das reine Geschäftsrisiko. Beispielsweise besteht bei der Distribution von Impfstoffen ein Risiko für die Qualität des Produktes in der korrekten Aufbewahrungstemperatur. Das zweite Risiko ist verbunden mit der Benutzung der IT-Systeme, zum Beispiel durch eine nicht korrekte Benutzung, durch technische Probleme des Systems selbst oder durch Hacker-Angriffe auf das System.
Testen, testen und nochmals testen
Alle Anforderungen an das System müssen getestet werden. Die Anforderungen werden auf verschiedenen Ebenen gestellt, somitsind auch Tests auf diesen Ebenen durchzuführen.
Auf der höchsten, der Benutzerebene, beschreibt der Benutzer, was das System leisten muss. Darunter die Anforderungen, die von Technikern (IT-Projektleiter) formuliert werden. Diese sind meistens in Module aufgeteilt: Networking, Datenbankzugriff, Benutzeroberfläche, benutzte Services und viele mehr. Geht man technisch dann weiter in die Tiefe, kommt man über Untermodule bis hin zur Programmierung, beschrieben zumeist von den Programmierern. Jeder dieser Spezifikationen entsprechen dann Tests.
Jeder Test benötigt einen Testplan, der den gesamten Test beschreibt. Dazu die Testfälle, die den einzelnen Tests entsprechen, und der abschließende Testreport, der die Ergebnisse der Tests zusammenfasst und einen Schluss aus den Testergebnissen zieht.
Hat man das System in der Testumgebung erfolgreich getestet, muss es in der Produktionsumgebung installiert und dort vom Endanwender getestet werden, um sicherzustellen, dass es auch dort konsistent die richtigen Ergebnisse liefert.
Cloudservices sind eine besondere Herausforderung an die Validierung computergestützter Systeme, vor allem was die Auditierung dieser Services seitens des Kunden und den eventuellen Speicherungsort der Daten anbetrifft. Im Moment beschäftigen sich Expertengruppen mit der Frage, inwieweit Cloudservices für GxP-kritische Geschäftsprozesse benutzt werden können und welche besonderen Maßnahmen zur Risikovorsorge getroffen werden müssen.
„Schützenhilfe“ bei Berufsverbänden
Es hängt von vielen Faktoren ab, welche Dokumentation produziert werden muss. In den GxP wird auf diese Fragestellung nicht genau eingegangen. Verschiedene Berufsverbände haben Empfehlungen verabschiedet, aus denen sich Teams, die computergestützte Systeme erstellen, schlaumachen können. Eine für die Pharmaindustrie weltweit anerkannte Richtlinie ist GAMP – „Good Automated Manufacturing Practices”. Sie wird vom internationalen non-profit Berufsverband ISPE herausgegeben.
ISPE – „International Society of Pharmaceutical Engineering“ – ist ein Berufsverband, der Guidelines für den Pharmabereich herausgibt, sowie auch eine Plattform darstellt, auf der man sich über Qualitätsthemen im Pharmabereich austauschen kann. Mitglieder im ISPE sind nicht nur Pharmafirmen, sondern auch private sowie öffentlich bestellte Inspektoren, Auditoren und Berater, die im Pharmaumfeld Dienstleistungen anbieten. Die GAMP-Richtlinien, die für diesen Artikel greifen, haben den Titel: „GAMP 5: Ein risikobasierter Ansatz für konforme GxP-computergestützte Systeme“ [ISPE].
Change-Management
Das Change-Management von computergestützten Systemen bedarf besonderer Aufmerksamkeit. Man unterscheidet hierbei zwei Arten von Changes, solche im Geschäftsprozess und solche in den IT-Systemen. Auch eine Veränderung im Geschäftsprozess kann eine Neuvalidierung der IT-Systeme notwendig mache.
Im Falle eines Change muss zuerst verstanden werden, wie tiefgreifend die Veränderung ist. Die Veränderung muss analysiert und eine Bewertung erstellt werden, inwieweit die Tests noch aussagekräftig sind. Die Entscheidung muss dokumentiert werden und die Dokumentation für spätere Konsultation oder auch Inspektionen, zum Beispiel durch Behörden, zur Verfügung stehen.
Ein wichtiger Aspekt in der Validierung von computergestützten Systemen ist der sogenannte „intended Use“. Das System ist nur dann in einem validierten Zustand, wenn es auch so benutzt wird, wie in den Nutzeranforderungen beschrieben.
Fazit
Was ist eigentlich der Unterschied im Aufwand zwischen der Entwicklung von computergestützten Systemen in GxP-kritischen Umgebungen und in Nicht-GxP-kritischen Umgebungen? Und was bedeutet dies konkret für den Softwaretest?
Der große Unterschied liegt im Ansatz. Nach den Nutzeranforderungen folgt im GxP-Umfeld sofort die Überlegung, wie kritisch der Geschäftsprozess und das dafür benutzte System sind. Nach dieser dokumentierten Überlegung beginnt der Entwicklungsprozess.
Im Entwicklungsprozess selbst wird im Fall „GxP-kritisch“ der Weg der Validierung eingeschlagen. Alle Anforderungen in allen Phasen werden dokumentiert und es wird eine Matrix angelegt, in der diese Anforderungen nachverfolgt werden können bis zu ihrer Implementierung und schließlich dem Test.
Im Fall „Nicht-GxP-kritisch“ muss das System zwar nicht validiert werden, das heißt jedoch keinesfalls, dass ohne Qualitätssicherung gearbeitet werden kann.
Ein zweiter Unterschied liegt, wie beschrieben, im Change-Management. Wird an einem validierten System oder an dem Geschäftsprozess, für das dieses validierte System benutzt wird, eine Änderung durchgeführt, so muss dieses System eventuell neu validiert werden. In einem Nicht-GxP-System ist dies nicht unbedingt notwendig.
Validierung bedeutet also dokumentierte Qualität des computergestützten Systems, und das kostet natürlich Ressourcen, auch in Form von Zeit. Bei Nicht-GxP-Systemen muss der Entwickler dann abwägen zwischen „Time to Market“ und der mindestens benötigten Qualität des Systems. Bei Systemen, die eventuell Konsequenzen für unsere Gesundheit haben, sollte klar sein, welchem Ziel der Vorrang gegeben wird.
Referenzen
[ISACA]
ISACA-Fachgruppe GXP,
https://www.isaca.de/de/fg-start/gxp
[ISPE]
ISPE GAMP 5 Guide,
https://ispe.org/publications/guidance-documents/gamp-5









